|
Metabarcoding and Metagenomics :
Research Article
|
|
Corresponding author: Nadine Graupner (nadine.graupner@uni-due.de)
Academic editor: Thorsten Stoeck
Received: 27 Jul 2017 | Accepted: 30 Aug 2017 | Published: 20 Sep 2017
© 2017 Nadine Graupner, Jens Boenigk, Christina Bock, Manfred Jensen, Sabina Marks, Sven Rahmann, Daniela Beisser
This is an open access article distributed under the terms of the Creative Commons Attribution License (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Citation:
Graupner N, Boenigk J, Bock C, Jensen M, Marks S, Rahmann S, Beisser D (2017) Functional and phylogenetic analysis of the core transcriptome of Ochromonadales. Metabarcoding and Metagenomics 1: e19862. https://doi.org/10.3897/mbmg.1.19862
|
|
Abstract
Background
Most protist lineages consist of members with diverging features e.g. different modes of nutrition and adaptations for life in different habitat types and climatic zones. The nutritional mode is particularly variable in chrysophytes and they are therefore an excellent model group to study the core genes and metabolic pathways of a functionally diverse lineage. The objective of our study is the identification of the joint genetic repertoire expressed in closely related chrysophytes as well as the extent of variation on species and strain level. Therefore, we investigated the transcriptomes of six strains belonging to four species of Ochromonadales. We performed analyses on metabolic pathway level as well as on sequence level.
Results
We could identify 1,574 core genes shared between all six investigated strains of Ochromonadales. Most of these core genes were affiliated with the primary metabolism. Phylogenetic analysis of 166 protein-coding core genes supported a close relation of Poteriospumella lacustris and Poterioochromonas malhamensis and resolved for more than 50% of investigated genes the relationship of strains affiliated with the species P. lacustris. Further, we found diverging phylogenetic patterns for genes interacting with the environment.
Conclusions
In Ochromonadales, a functionally diverse lineage, the core transcriptome represents only a minor part of the individual transcriptomes. But this small fraction of genes comprises the basal metabolism essential for life in several protist lineages. Phylogenetic analyses of these genes indicate a similar degree of conservation as observed for genes coding for ribosomal proteins.
Keywords
Chrysophyceae, protist, expressed sequence tags (EST), evolutionary ecology, phylotranscriptomics, core genes, metabolic pathways
Introduction
Organisms and their genomes evolved under persistently fluctuating ecological and evolutionary pressures. As a consequence, eukaryotic genomes and their gene content vary considerably in size ranging from 2,000 to 35,000 genes (
Previous investigations of core genes aimed at various research topics spanning from the identification of the minimal gene set necessarily for cellular life (
Further, we ask the question to what extent do gene phylogenies reflect the history, i.e. phylogeny, of the organism and to what extent are these pattern concealed by ecological adaptation. Recent studies (
We used six strains affiliated with four species within the order Ochromonadales (Chrysophyceae), i.e. Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11), which were part of the overarching transcriptome study of
Material and methods
Cultivation of strains and RNA isolation
We focused on strains affiliated with the order Ochromonadales. For our study we used six strains affiliated with four species: Poteriospumella lacustris, Poterioochromonas malhamensis, Spumella vulgaris and Pedospumella encystans. For further information regarding origin, mode of nutrition and culturing conditions see Table
| Poteriospumella lacustris | Poterioochromonas malhamensis | Spumella vulgaris | Pedospumella encystans | |
| Strain | JBC07, JBM10, JBNZ41 | DS | 199hm | JBMS11 |
| 18S clade | C3 | C3 | C2 | C1 |
| Geographical origin | China, Austria, New Zealand | Austria | Arctic | Austria |
| Habitat origin | freshwater | freshwater | freshwater | soil |
| Mode of nutrition | heterotroph | mixotroph | heterotroph | heterotroph |
| Media | IB + 3g/l nutrient broth, soytone & yeast extract | IB + 3g/l nutrient broth, soytone & yeast extract | IB + bacteria (Listonella pelagia PG5) | IB + bacteria (Listonella pelagia PG5) |
| Temperature | 15°C | 15°C | 15°C | 15°C |
| Light:dark-cycle | 16 : 8 | 16 : 8 | 16 : 8 | 16 : 8 |
| Illumination | 75 - 100 µE | 75 - 100 µE | 75 - 100 µE | 75 - 100 µE |
Sequencing, assembly and annotation
The transcriptome sequences were generated in an overarching study of 18 chrysophyte strains, published in
Base quality of raw sequence reads was checked using the FastQC software (v0.10.1;
Within the KEGG database genes are associated with orthologous groups and thus assigned to KEGG Orthology (KO) identifiers. In the following we use the term gene for the annotated orthologous gene of the considered transcript.
Comparative analysis of core, shared and exclusive genes
The assigned KO identifiers were used to determine the core transcriptome, constituted by the intersection of the KO identifier sets between all strains, shared transcripts between several species and exclusive transcripts of single strains using the R package Vennerable (3.0;
Pathway analysis
Orthologous genes (KOs) assignable to KEGG pathways of the KEGG BRITE functional hierarchy level A, B and C (
Expression analysis
Gene expression counts of all six strains were compared. Therefore, transcript expression values were obtained with the tool eXpress (v1.3.1;
Phylogenetic analysis
To create sequence alignments the pairwise alignments to KEGG gene sequences were used as a reference. All transcripts aligning to the same gene were truncated to the minimum overlapping region of at least 100bp and combined in a fasta file. The sequence alignments were constructed with the MAFFT software (7.164b;
Data resources
European Nucleotide Archive (ENA) under accession number PRJEB13662
Results
General aspects - Transcriptome sizes and functional assignment
Sequencing of the chrysophyte strains resulted in 13.8 to 19.4 million read pairs, which were assembled into 24,783 to 58,003 transcripts (Table
Overview statistics of Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11) including transcriptome size, assembly quality and annotation.
| P. lacustris (JBC07) | P. lacustris (JBM10) | P. lacustris (JBNZ41) | P. malhamensis (DS) | S. vulgaris (199hm) | P. encystans (JBMS11) | |
| No. read pairs (million) | 13.8 | 19.4 | 15.8 | 18.8 | 13.9 | 14.2 |
| Reads after quality control (%) | 92.5 | 91.7 | 92.4 | 94.3 | 92.4 | 93 |
| No. transcripts | 24,783 | 26,330 | 27,754 | 39,537 | 58,003 | 40,532 |
| N50 | 1,155 | 1,246 | 1,275 | 1,405 | 983 | 1,077 |
| Remapped reads (%) | 97.3 | 97 | 91.6 | 95.1 | 89.5 | 86.1 |
| Estimated no. of protein-coding genes | 20,441 | 20,515 | 21,629 | 30,189 | 38,883 | 28,497 |
| No. KEGG hits (E-value < 10-5) | 6,619 | 6,784 | 7,025 | 9,556 | 10,711 | 9,893 |
| No. unique KEGG orthologs | 2,248 | 2,265 | 2,265 | 2,620 | 2,694 | 2,652 |
| No. KEGG orthologs assignable to pathways | 1,367 | 1,389 | 1,378 | 1,599 | 1,635 | 1,591 |
| No. assigned KEGG pathways | 243 | 246 | 244 | 247 | 259 | 257 |
Estimates based on the number of Trinity components yielded a maximum of 20,441 to 38,883 genes. The transcriptomes of the three strains of Poteriospumella lacustris displayed a similar number of estimated genes (20,441 to 21,629 Trinity components) and similar functional annotations despite diverging sequencing depths. This finding and the completeness of several KEGG modules (main reaction steps in KEGG pathways), e.g. glycolysis, citrate cycle, oxidative phosphorylation and nucleotide biosynthesis, denoted a sufficient sequencing depth in all samples.
Between 45.2% and 56.7% of all predicted genes (Trinity components) could be assigned to a gene in the KEGG database. The removal of redundant hits resulted in 2,249 to 2,695 unique KEGG orthologous genes of which 1,367 to 1,635 could be assigned to 243 to 259 pathways (Table
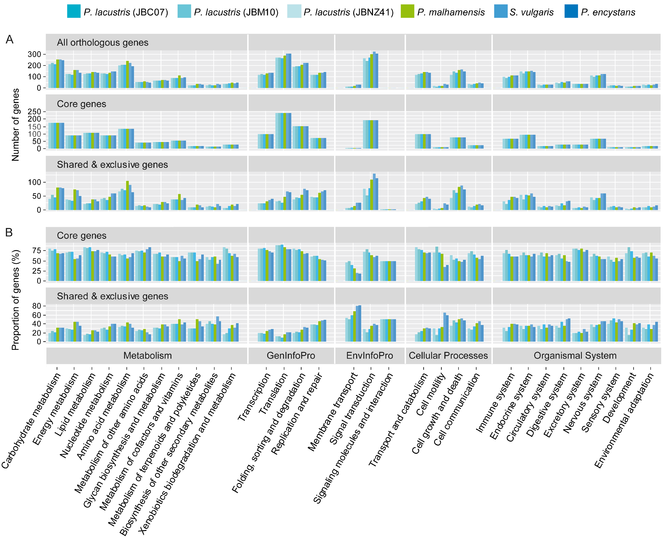
Functional assignment of non-redundant KEGG orthologous genes of Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11) to 31 pathway groups of the KEGG categories “Metabolism”, “Genetic Information Processing” (GenInfoPro), “Environmental Information Processing” (EnvInfoPro), “Cellular Processes” and “Organismal Systems”. A All functional hits per pathway group and strain were summarized for the whole transcriptomes, for the core transcriptome of all six strains and for the shared and exclusive genes. B The percentage proportion of genes per pathway group and strain was calculated for the core genes and the shared and exclusive genes.
Comparison of strains
A PCoA based on presence-absence data of KOs clearly separated all species (Fig.
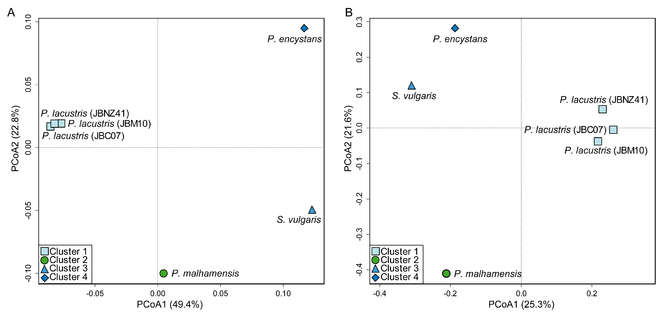
PCoA based on all identified KEGG orthologous genes of Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11). A PCoA based on the presence-absence of the respective genes. B PCoA based on the relative expression level of the respective genes.
Genes that were responsible for the grouping of strains were present in several but not in all species (around 800 genes) or exclusive to one species (181 to 307 genes). Poteriospumella lacustris had the lowest number and Spumella vulgaris the highest number of exclusive genes. Most of these partly shared and exclusive genes were affiliated with “Signal transduction”, “Amino acid metabolism”, “Cell growth and death”, “Carbohydrate metabolism”, “Energy metabolism” and “Replication & repair” (Fig.
The pairwise comparison showed that the percentage of shared KEGG orthologous genes (Fig.
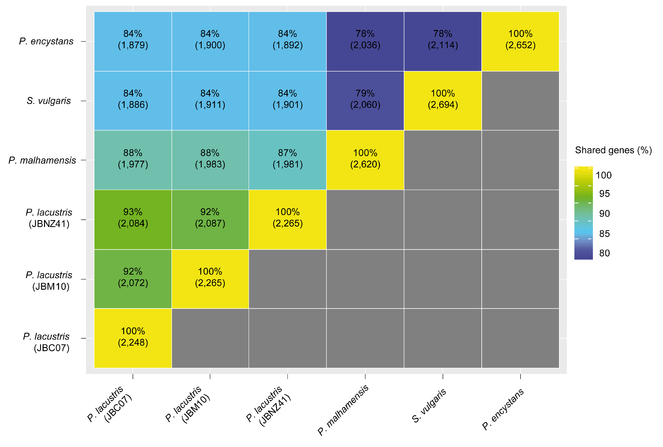
Pairwise comparison of the proportion of shared KEGG orthologous genes of Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11). The values in each matrix element are provided as the percentage of shared KEGG orthologous genes in relation to the number of KEGG orthologous genes in the smaller transcriptome. The absolute number of shared KEGG orthologous genes is given in brackets.
Sequence comparisons based on all transcripts (instead of the annotated ones only) revealed that the majority of transcripts was exclusive to one strain. However, the strains of Poteriospumella lacustris shared approximately 50% of their transcripts with each other also indicating their close relation. The number of core transcripts was comparable to those of the annotated part.
Core transcriptome
The core transcriptome derived from the annotated part of the transcriptomes of the investigated strains of Ochromonadales comprised 1,574 KEGG orthologous genes. Of these, we were able to assign 1,017 to one or more KEGG pathways (Fig.
The core transcriptome comprised a large number of genes involved in basic cell metabolism: Roughly 38% of the genes of the core transcriptome were assigned to “Metabolism” and 27% of the genes were assigned to “Genetic Information Processing” (Fig.
Genes of the core transcriptome that were highly expressed in all strains were affiliated with the KEGG category “Genetic Information Processing” coding for various ribosomal proteins of the large and small subunit of ribosomes and the elongation factor affiliated with RNA transport. But also genes like ubiquitin c, heat shock proteins, calmodulin and solute carrier proteins affiliated with various signaling pathways as well as the F-type H+-transporting ATPase affiliated with the oxidative phosphorylation were highly expressed.
Phylogenetic inference
Phylogenetic analyses were performed for 166 orthologous genes (Suppl. material
In topology A (Fig.
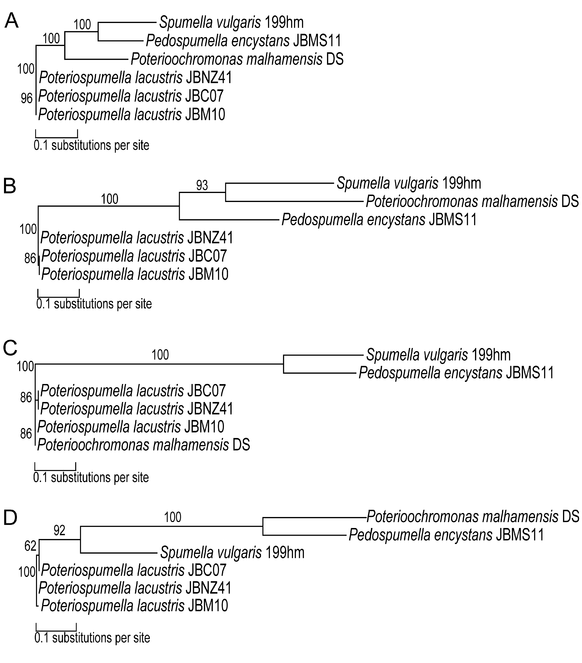
Main topologies (unrooted phylogenetic trees; values at the nodes indicate statistical support >50% estimated by maximum likelihood method with 1,000 replicates) of 166 investigated KEGG orthologous genes of the core transcriptome of Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11). For each topology one gene is exemplarily illustrated. A most frequent tree topology (obtained for 76 genes), B second most frequent tree topology (obtained for 36 genes), C third most frequent tree topology (obtained for 19 genes), and D fourth most frequent tree topology (obtained for 13 genes).
The phylogenetic relation between the three strains of Poteriospumella lacustris was resolved by approximately 58% of the investigated genes; approximately 53% of these indicated the closest relationship between the strains JBC07 and JBNZ41. Further, approximately 36% of these genes indicated a close relationship between strain JBM10 and Poterioochromonas malhamensis.
The affiliation to a distinct pathway is known for 117 of the genes included in the phylogenetic analysis (Fig.
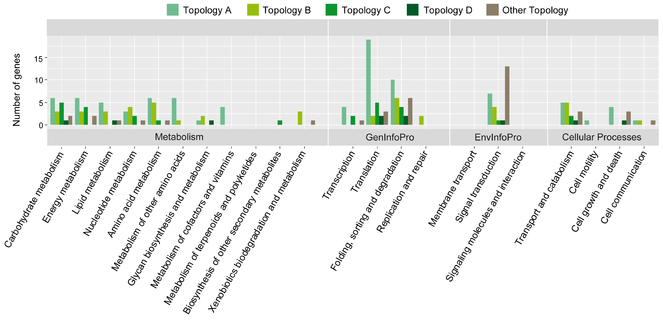
Functional assignment of core genes used for phylogeny analyses of Poteriospumella lacustris (JBC07, JBM10, JBNZ41), Poterioochromonas malhamensis (DS), Spumella vulgaris (199hm) and Pedospumella encystans (JBMS11). The KEGG category “Organismal Systems” was not illustrated as no core genes were affiliated to this category.
Discussion
The chrysophyte core transcriptome – the essential minority
The comparative transcriptomic approach allowed insights into the genetic repertoire transcribed by Ochromonadales showing that the transcriptomes differed considerably between strains. Between 20,441 and 38,883 estimated protein coding genes were identified. This is in the lower and mid-range of previously reported values for gene estimates based on transcriptomes (
The majority of genes could not be affiliated with a distinct function.
Accordingly, roughly 65% of the identified orthologous genes are part of the core transcriptome in our study. A significant fraction of these core genes, was affiliated with the primary metabolism, reflecting the general importance of these metabolic pathways irrespective of phylogeny, nutritional mode and origin. They were affiliated with metabolic pathways including translation and ribosomal biogenesis, transcription and protein processing as well as with pathways affiliated with “Carbohydrate metabolism”, “Lipid metabolism”, “Nucleotide metabolism”, “Amino acid metabolism” and “Signal transduction”. This corresponds well with transcriptomic studies of other taxa which revealed a similar number and affiliation of genes of the core transcriptome with metabolic pathways (prymnesiophytes 1,433 core genes:
Gene function interferes with phylogenetic signals in tree topologies
We calculated alignment-based phylogenetic trees of the six investigated strains for 166 core genes. The analyses resulted in four different tree topologies. The most frequent and the third frequent topology confirmed the close relation of Poterioochromonas malhamensis and Poteriospumella lacustris inferred from SSU rRNA gene sequences (both species are members of the C3-clade:
Furthermore, our analyses helped to resolve the relationship between closely related strains affiliated with one species. More than 50% of the investigated protein-coding genes showed sequence variations between the three strains JBC07, JBM10 and JBNZ41, which all belong to the species Poteriospumella lacustris. Earlier studies of these strains based on a multigene phylogeny of the protein-coding genes alpha-tubulin, beta-tubulin and actin (
Conclusions
The present study reveals the interplay of functionality and phylogeny of the core transcriptome of Ochromonadales. We could demonstrate that the core transcriptome of Ochromonadales with its 1,574 genes represents only a small proportion of the transcriptomes but it comprises the genes affiliated with the primary metabolism. We assume that roughly 1,400 genes represent the basic “active” genetic repertoire of various protist lineages. Furthermore, we performed phylogenetic analyses of 166 protein-coding core genes. Most of the investigated genes coding for ribosomal genes or metabolism confirmed the close relation of Poterioochromonas malhamensis and Poteriospumella lacustris known from SSU rRNA gene phylogenies. Genes interacting with the environment largely show diverging phylogenetic patterns presumably due to a stronger impact of ecological selection pressures. Furthermore, we demonstrated the strength of comparative transcriptomics for the analysis of intraspecific and interspecific variation. Both, orthologous gene content analysis (PCoA) and phylogenetic analyses for several genes lead to congruent results of the relationship of Ochromonadales supporting the robustness of our results.
Acknowledgements
We thank the projects DFG Projekt BO 3245/17 and BO 3245/14 for financial support. Further, we thank Susann Chamrad for technical assistance.
Funding program
DFG Projekt BO 3245/17 and BO 3245/14.
Conflicts of interest
The authors declaire no conflict of interests.
References
- Phylogenetic analysis of the SSU rRNA from members of the Chrysophyceae.Protist150(March):71‑84. https://doi.org/10.1016/S1434-4610(99)70010-6
- FastQC: A quality control tool for high throughput sequence data.https://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- The genome of the diatom Thalassiosira pseudonana: Ecology, evolution, and metabolism.Science306(5693):79‑86. https://doi.org/10.1126/science.1101156
- A kingdom-level phylogeny of eukaryotes based on combined protein data.Science290(5493):972‑977. https://doi.org/10.1126/science.290.5493.972
- Comprehensive transcriptome analysis provides new insights into nutritional strategies and phylogenetic relationships of chrysophytes.PeerJ5https://doi.org/10.7717/peerj.2832
- High diversity of the 'Spumella-like' flagellates: An investigation based on the SSU rRNA gene sequences of isolates from habitats located in six different geographic regions.Environmental Microbiology7(5):685‑697. https://doi.org/10.1111/j.1462-2920.2005.00743.x
- The Phaeodactylum genome reveals the evolutionary history of diatom genomes.Nature456(7219):239‑244. https://doi.org/10.1038/nature07410
- Probing the evolution, ecology and physiology of marine protists using transcriptomics.Nature Reviews Microbiology15(1):6‑20. https://doi.org/10.1038/nrmicro.2016.160
- Molecular Biology: Principles and Practice.W. H. Freeman and Company, New York..
- Emerging Diversity within Chrysophytes, Choanoflagellates and Bicosoecids Based on Molecular Surveys.Protist162(3):435‑448. https://doi.org/10.1016/j.protis.2010.10.003
- Mining from transcriptomes: 315 single-copy orthologous genes concatenated for the phylogenetic analyses of Orchidaceae.Ecology and Evolution5(17):3800‑3807. https://doi.org/10.1002/ece3.1642
- Transcriptome sequencing of three Pseudo-nitzschia species reveals comparable gene sets and the presence of Nitric Oxide Synthase genes in diatoms.Scientific reports5(April):12329‑12329. https://doi.org/10.1038/srep12329
- Transcriptomics: Advances and approaches.Science China-Life Sciences56(10):960‑967. https://doi.org/10.1007/s11427-013-4557-2
- Why highly expressed proteins evolve slowly.Proc Natl Acad Sci USA102(40):14338‑43. https://doi.org/10.1073/pnas.0504070102
- Microbial community structure and its functional implications.Nature459(7244):193‑9. https://doi.org/10.1038/nature08058
- Complete Chemical Synthesis, Assembly, and Cloning of a Mycoplasma genitalium Genome.Science319(5867):1215‑1220. https://doi.org/10.1126/science.1151721
- Determination of the Core of a Minimal Bacterial Gene Set Determination of the Core of a Minimal Bacterial Gene Set.Microbiology and Molecular Biology Reviews68(3):518‑537. https://doi.org/10.1128/MMBR.68.3.518
- Full-length transcriptome assembly from RNA-Seq data without a reference genome.Nature Biotechnology29(7):644‑52. https://doi.org/10.1038/nbt.1883
- Small but Manifold – Hidden Diversity in “Spumella-like Flagellates”.Journal of Eukaryotic Microbiology63(4):419‑439. https://doi.org/10.1111/jeu.12287
- The genetic core of the universal ancestor.Genome research13(3):407‑412. https://doi.org/10.1101/gr.652803
- The first de novo transcriptome of pepino (Solanum muricatum): assembly, comprehensive analysis and comparison with the closely related species S. caripense, potato and tomato.BMC Genomics17(1). https://doi.org/10.1186/s12864-016-2656-8
- Comparison of the Yeast Proteome to Other Fungal Genomes to Find Core Fungal Genes.Journal of Molecular Evolution60(4):475‑483. https://doi.org/10.1007/s00239-004-0218-1
- KEGG: Kyoto Encyclopedia of Genes and Genomes.Nucleic Acids Research28(1):27‑30. https://doi.org/10.1093/nar/28.1.27
- KEGG: new perspectives on genomes, pathways, diseases and drugs.Nucleic Acids Research45https://doi.org/10.1093/nar/gkw1092
- MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability.Molecular biology and evolution30(4):772‑780. https://doi.org/10.1093/molbev/mst010
- Comparative transcriptome analysis of four prymnesiophyte algae.Plos One9(6). https://doi.org/10.1371/journal.pone.0097801
- Comparative genomics, minimal gene-sets and the last universal common ancestor.Nature Reviews Microbiology1(2):127‑136. https://doi.org/10.1038/nrmicro751
- Proteinortho: Detection of (Co-)orthologs in large-scale analysis.BMC Bioinformatics12(1):124‑124. https://doi.org/10.1186/1471-2105-12-124
- Transcriptome sequencing and phylogenomic resolution within Spalacidae (Rodentia).BMC Genomics15:32‑32. https://doi.org/10.1186/1471-2164-15-32
- Gene expression characterizes different nutritional strategies among three mixotrophic protists.Fems Microbiology Ecology92(7):1‑11. https://doi.org/10.1093/femsec/fiw106
- Cutadapt removes adapter sequences from high-throughput sequencing reads.EMBnet.journal17(1):10‑12. https://doi.org/10.14806/ej.17.1.200
- GeneDoc: a tool for editing and annotating multiple sequence alignments.http://www.psc.edu/biomed/genedoc/.
- vegan: Community ecology package.R package version 2.4.-1.URL: https://CRAN.R-project.org/package=vegan
- Robust Dinoflagellata phylogeny inferred from public transcriptome databases.J Phycol53(3):725‑729. https://doi.org/10.1111/jpy.12529
- Draft genome sequence and genetic transformation of the oleaginous alga Nannochloropis gaditana.Nat Commun3https://doi.org/10.1038/ncomms1688
- Streaming fragment assignment for real-time analysis of sequencing experiments.Nat Meth10(1):71‑73. https://doi.org/10.1038/nmeth.2251
- phangorn: phylogenetic analysis in R.Bioinformatics27(4):592‑593. https://doi.org/10.1093/bioinformatics/btq706
- Scale evolution in Paraphysomonadida (Chrysophyceae): Sequence phylogeny and revised taxonomy of Paraphysomonas, new genus Clathromonas, and 25 new species.European Journal of Protistology50(5):551‑592. https://doi.org/10.1016/j.ejop.2014.08.001
- Metagenomic microbial community profiling using unique clade-specific marker genes.Nat Meth9(8):811‑814. https://doi.org/10.1038/nmeth.2066
- Developments in the taxonomy of silica-scaled chrysophytes - from morphological and ultrastructural to molecular approaches.Nordic Journal of Botany31(4):385‑402. https://doi.org/10.1111/j.1756-1051.2013.00119.x
- Multigene phylogenies of clonal Spumella-like strains, a cryptic heterotrophic nanoflagellate, isolated from different geographical regions.International Journal of Systematic and Evolutionary Microbiology58(3):716‑724. https://doi.org/10.1099/ijs.0.65310-0
- TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses.BMC Bioinformatics11https://doi.org/10.1186/1471-2105-11-7
- Transcriptome-wide evolutionary analysis on essential brown algae (Phaeophyceae) in China.Acta Oceanologica Sinica33(2):13‑19. https://doi.org/10.1007/s13131-014-0436-3
- Venn diagrams in R with the Vennerable package.https://rdrr.io/rforge/Vennerable/f/inst/doc/Venn.pdf.
- Comparative analysis on transcriptome sequencings of six Sargassum species in China.Acta Oceanologica Sinica33(2):37‑44. https://doi.org/10.1007/s13131-014-0439-0
- Phylotranscriptomic analysis of the origin and early diversification of land plants.Proceedings of the National Academy of Sciences of the United States of America111(45):4859‑68. https://doi.org/10.1073/pnas.1323926111
- ggplot2: elegant graphics for data analysis.Springer-Verlag, New York.
- Origin of land plants: Do conjugating green algae hold the key?BMC Evolutionary Biology11(1):104‑104. https://doi.org/10.1186/1471-2148-11-104
- Analysis of pan-genome to identify the core genes and essential genes of Brucella spp.Molecular Genetics and Genomics291(2):905‑912. https://doi.org/10.1007/s00438-015-1154-z
- RAPSearch2: a fast and memory-efficient protein similarity search tool for next-generation sequencing data.BIOINFORMATICS APPLICATIONS NOTE28(1):125‑126. https://doi.org/10.1093/bioinformatics/btr595
Supplementary material
KEGG orthologous genes of the core trancriptome of the herein investigated Ochromonadales (Poteriospumella lacustris strains JBC07, JBM10, JBNZ41; Poterioochromonas malhamensis DS; Spumella vulgaris 199hm; Pedospumella encystans JBMS11) used for phylogenetic analyses.